How many cells to sample?
Having relatively limited experience in the world of single cells, one question that stumped me was how many cells does one need to sequence? Given my background in instrumentation, my experience in single cell world was always limited by the actual hardware. More is always better from an instrument standpoint!
Turns out, things are never as simple as they seem: sequencing cost is the limiting factor, especially in single-cell DNA-seq.
Here is a great infographic of capabilities of scDNA-seq (Fig A-C). What really answers my question is Fig D, which neatly summarizes the application-specific requirements for throughput vs genome coverage (aka sequencing costs).
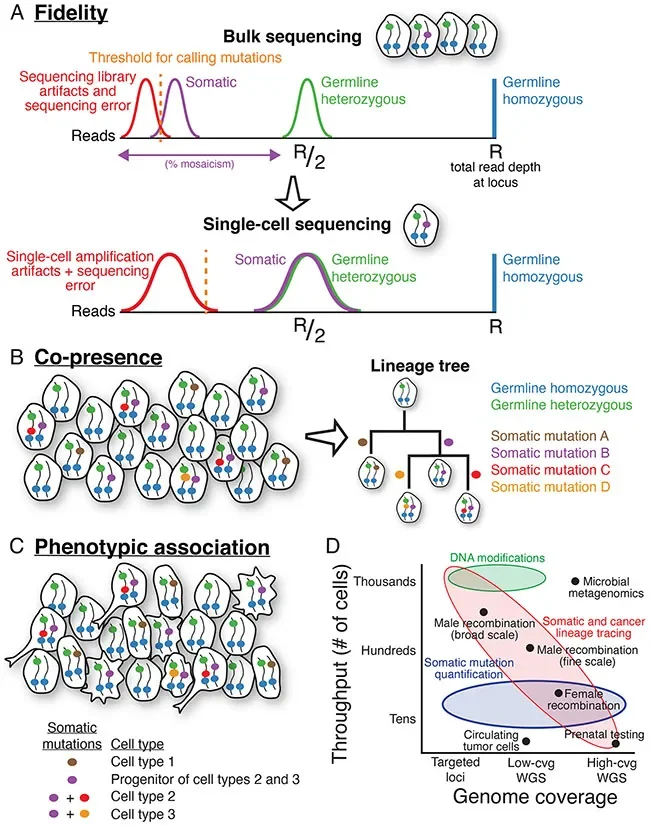
In summary, there is no one throughput fits all for scDNA-seq. Higher coverage requires more money, and high throughput x high coverage might not be necessary for some samples. E.g. to detect the influence of segmental CNVs in diseases for tissues with abundant subclones, you might need to sequence 0.02x to 0.05x depth per cell (equivalent to 400K-1M reads), which isn’t a lot of money with higher throughput in the thousands cell range currently. In preimplantation genetic testing (PGT), as little cells need to be removed as possible (currently the safest practice is the trophectoderm biopsy of a day-5 developing embryo), and most tend to sequence at 30x read depth. For circulating tumour cells (CTCs), there is a limited number of samples that can be obtained from patients. From the same review, 27% of samples from prostate cancer patients did not have detectable CTCs, and a median of 7 CTCs were found in remaining samples.
This is now a statistical question for how many cells do I need to sequence in order to detect the subpopulation that I want, at a given confidence.
The problem becomes this:
true frequency —> sampled count —> estimated frequency —> sample size calculation
I found this article by Navin that essentially explains this problem and why there is some difficulty in estimation these factors (Box 1),
Diversity of underlying population
Sensitivity required to detect rare clones (usually around 1% frequency)
Technical variability of single cell sequencing method.
It also points to various statistical methods from ecology and population genetics to help estimate sample size, mainly binomial distribution.
Satija lab has this great calculator: https://satijalab.org/howmanycells/
Navin’s lab has expanded the bionomial to multinomial distribution with SCOPIT: https://alexdavisscs.shinyapps.io/scs_power_multinomial/ (paper here: https://pubmed.ncbi.nlm.nih.gov/31718533/)
SCOPIT has a prospective and retrospective tab. For the retrospective method, you can use it to plan for an experiment if you don’t have information about the frequencies for each type of cell but have previous experiment data. Prospective is when you know the frequency of each cell type (which you may probably not know). It also allows you to input additional subpopulations with different frequencies, which the Satija calculation does not account.
A concrete example following this logic:
true frequency —> sampled count —> estimated frequency —> sample-size calculation
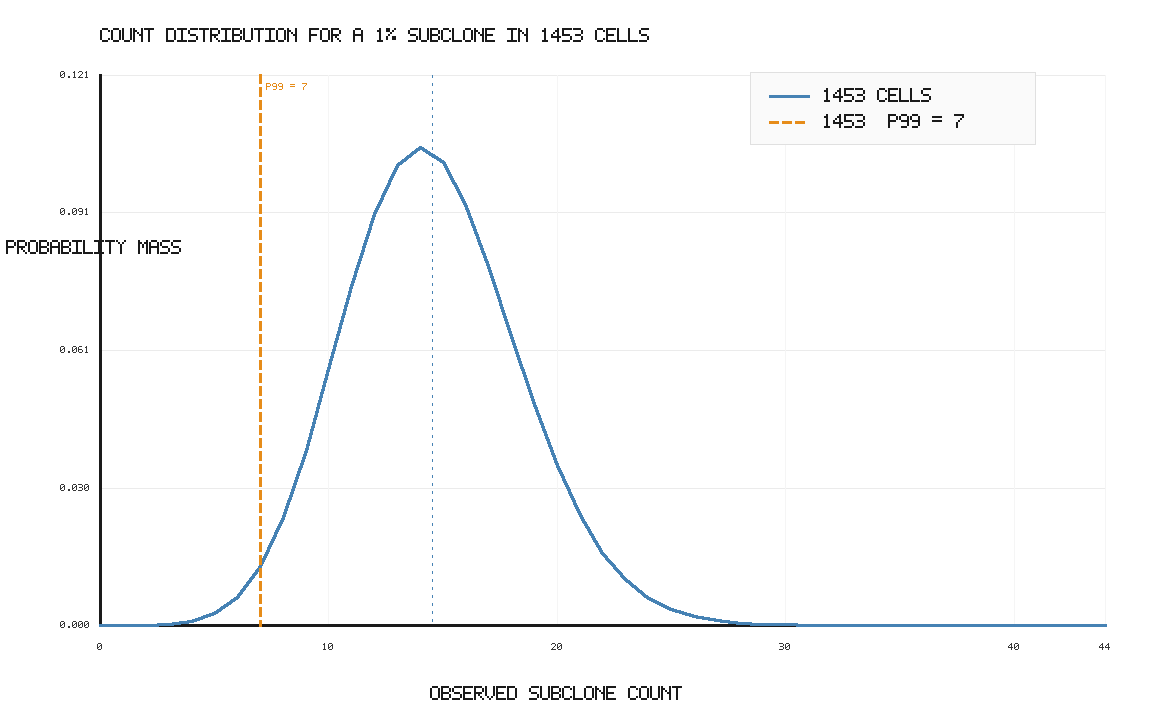
Let’s assume the true subclone frequency (f) = 1%.
If I dispense 1000 cells, then the number from that subclone is random:
X ~ Binomial (1000,0.01)
Biologically this means that if the tissue truly contains 1% of that subclone, then I will average about 1000*0.01 = 10 cells. But there is always some deviation, meaning that if I repeat multiple times, I may get 7 or 11 by chance (aka 0.7% estimated frequency, then 1.1% the next time), which is not true frequency, just estimated frequency.
The next step is to define what enough cells mean. Here I want 7 cells from one subclone. What is the smallest number of total cells (including my 7 cells) such that I have a % chance of observing c = 7 cells from this one subclones? I defined my % chance as 99%, which means:
X ~ Binomial(n, 0.01) and the success condition is P(X >= 7) >= 0.99
Since it’s easier to calculate failure probability, it becomes 1 - P(X <= 6) >= 0.99.
The binomial probability of observing exactly k=7 subclone cells is:
P(X = k) = choose(n, k) * f^k * (1 - f)^(n-k)
So the probability of failing to get 7 cells is:
P(X <= 6) = P(X = 0) + P(X = 1) + ... + P(X = 6)
or: P(X <= 6) = sum from k = 0 to 6 of choose(n, k) * 0.01^k * 0.99^(n-k)
The calculator then searches over possible values of n until it finds the first value where: 1 - sum from k = 0 to 6 of choose(n, k) * 0.01^k * 0.99^(n-k) >= 0.99
For this example, the answer is approximately n = 1453 cells.
This is higher than the required 7 cells, but that is the point: to be 99% confident that you get at least 7 cells, the average has to be well above 7. If the average were only 7, then about half of experiments would fall below 7 due to sampling noise.
So the full logic is:
true frequency = 1%
sampled count = random, e.g. 10 out of 1000
estimated frequency = 10 / 1000 = 1%
desired minimum captured cells = 7
desired probability of success = 99%
calculator finds n such that P(X >= 7) >= 0.99
required sample size ≈ 1453 cells
The probability mass function will look something like this:
I also tried to think of it in another way. Since I am an instrumentation person and the throughout is limited by how many cells is in my instrument throughput, I was wondering if one could estimate how many cells from one subclone I can get if limited by dispensing a specific number of cells into a plate e.g. after FACS.
From poking around, if I want to get at least 2 cells from one subpopulation with 95% probability, I would need 473 cells. With 99% probability then I would need 662 cells! That’s a lot of plates to sort cells into a 384-well plate and run scDNA-seq in each well…that’s why I care about increasing throughput from an instrument perspective.
So if I could increase my instrument throughput from 1000 cells to 3000 cells:
From an instrument perspective, throughput determines how far into the sampling distribution we can push. At 1% subclone frequency, 1000 cells gives an expected 10 subclone cells, but only guarantees at least 3 cells with 99% probability. Increasing throughput to 3000 cells raises the expected count to 30 and raises the 99% lower-bound count to about 18 cells.
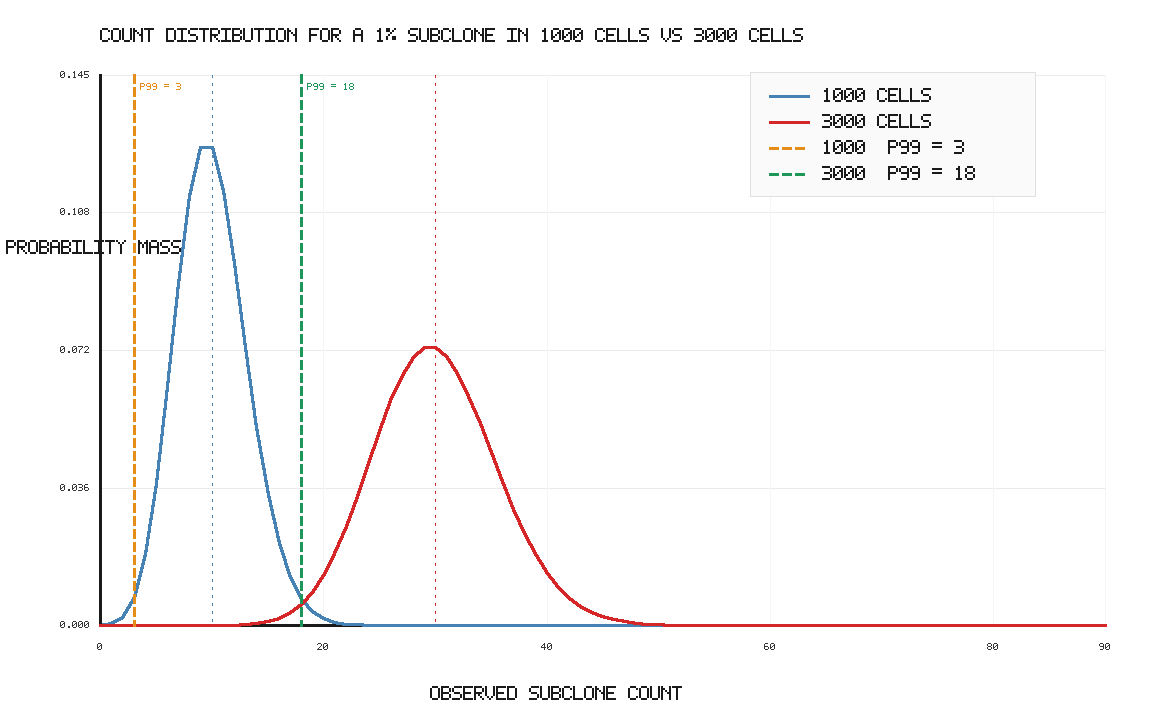
Another way to visualize the throughput limitation:
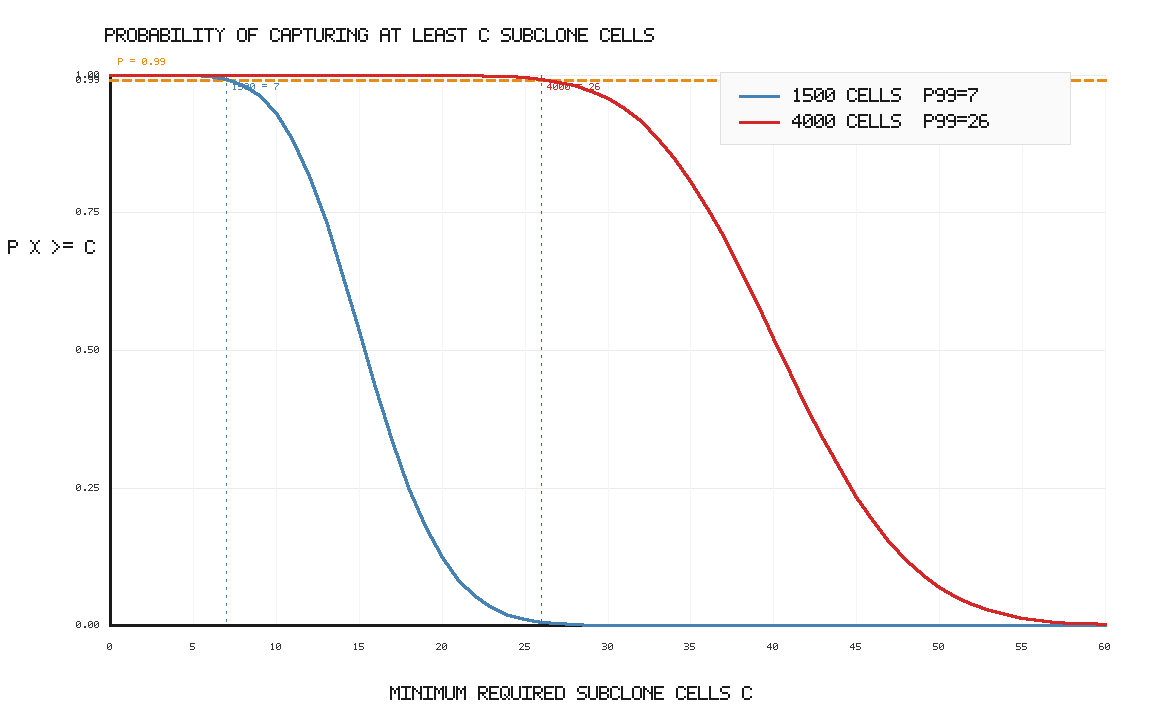
Clearly there are a ton of single cell instruments out there with much higher throughput than my hypothetical 1000 cells, with 10x Chromium leading the pack. For single plex, you could get 160K cells/chip (20K cells/sample x 8 lanes). For plate-based multiplexing, one could get 20K cells/sample and 8M cells/chip.
Hopefully this bridges the biology, the probability calculation, and why instrument throughput matters to me!